Dassanayake Lab
COMPARATIVE
FUNCTIONAL
& EVOLUTIONARY
GENOMICS

Schrenkiella parvula Genome V2.2
NCBI taxonomy id: 98039
Phytozome id: 574
Overview
Schrenkiella parvula is an extremophyte model that thrives under salt, drought, flooding, chilling, high light, and heat stresses while being able to grow in low nitrogen and phosphorus soils often enriched in other elements toxic to most plants. All plants respond to environmental stress, but most plants fall short as study systems because they fail to sustain growth at high levels of environmental stresses. Extremophytes are unique in their biology and exhibit adaptations to sustain growth under diverse and extreme abiotic stresses and thus show great promise for discovering novel genetic mechanisms underlying plant adaptations to environmental stresses. S. parvula is an ideal genetic model that can grow under multiple environmental stresses while maintaining significant growth; can be easily grown in controlled environments; and has the foundational resources established for functional genomic studies (Dassanayake et al., 2011; Oh et al., 2012; Wang et al., 2019). It is primarily distributed around salt lakes in the Irano-Turanian floristic region (Uzilday et al., 2015; Hajiboland et al., 2018).
S. parvula is in the plant family, Brassicaceae. Brassicaceae is among the most genomic-resource rich plant families (Koenig & Weigel, 2015), that provides an unprecedented resource to study how distinct environments have shaped closely-related species into diverse lifestyles. This family not only contains the model plant Arabidopsis thaliana with the most genomic information available for any plant species, but also includes a large number of crops (Kiefer et al., 2014). Therefore, S. parvula presents a prime model to identify novel stress tolerance mechanisms that are missing or difficult to study in A. thaliana and other stress-sensitive crop models in a comparative framework. S. parvula is the first publicly available extremophyte model genome (Dassanayake et al., 2011) and represent a well-recognized resource for understanding genetic mechanisms and novel gene functions related to stress adaptations (Dittami & Tonon, 2012; Oh et al., 2012; Kazachkova et al., 2018).
S. parvula has been known as Arabidopsis parvula, Thellungiella parvula, and Eutrema parvulum in the recent past (‘The Plant List’, 2013). It is important to note that multiple publications on this species may not provide all of its former names or may still use a former name without referring to its currently accepted name, S. parvula. It is also occasionally referred to as salt cress due to its former placement in the genus Thellungiella and its recognition as a halophyte model (Whited, 2015). Despite the distinct choice of nomenclature used by different groups, all publications converge on the use of a single species with clear adaptations observed for abiotic stresses and the majority of the genetic studies published have used the ecotype, Lake Tuz, Turkey. Notably, S. parvula represents the most salt-tolerant species (based on soil NaCl LD50) among wild relatives of A. thaliana (Orsini et al., 2010).
The primary goal of this genome project was to provide a highly contiguous assembly at the chromosomal-scale for S. parvula as much as possible given the sequencing platforms available in 2010 and further update it with a curated annotation to facilitate our fundamental understanding of gene models from an extremophyte model that can provide insights into plant stress adaptations. Therefore, we invite the plant community interested in using this resource to expand their research using it at the whole genome to single gene level as needed. Identifying those extremophyte genes and their functional regulatory networks would allow us to strategize how best we can adapt the functions of stress adaptive genes into our crops to better prepare for a changing climate.
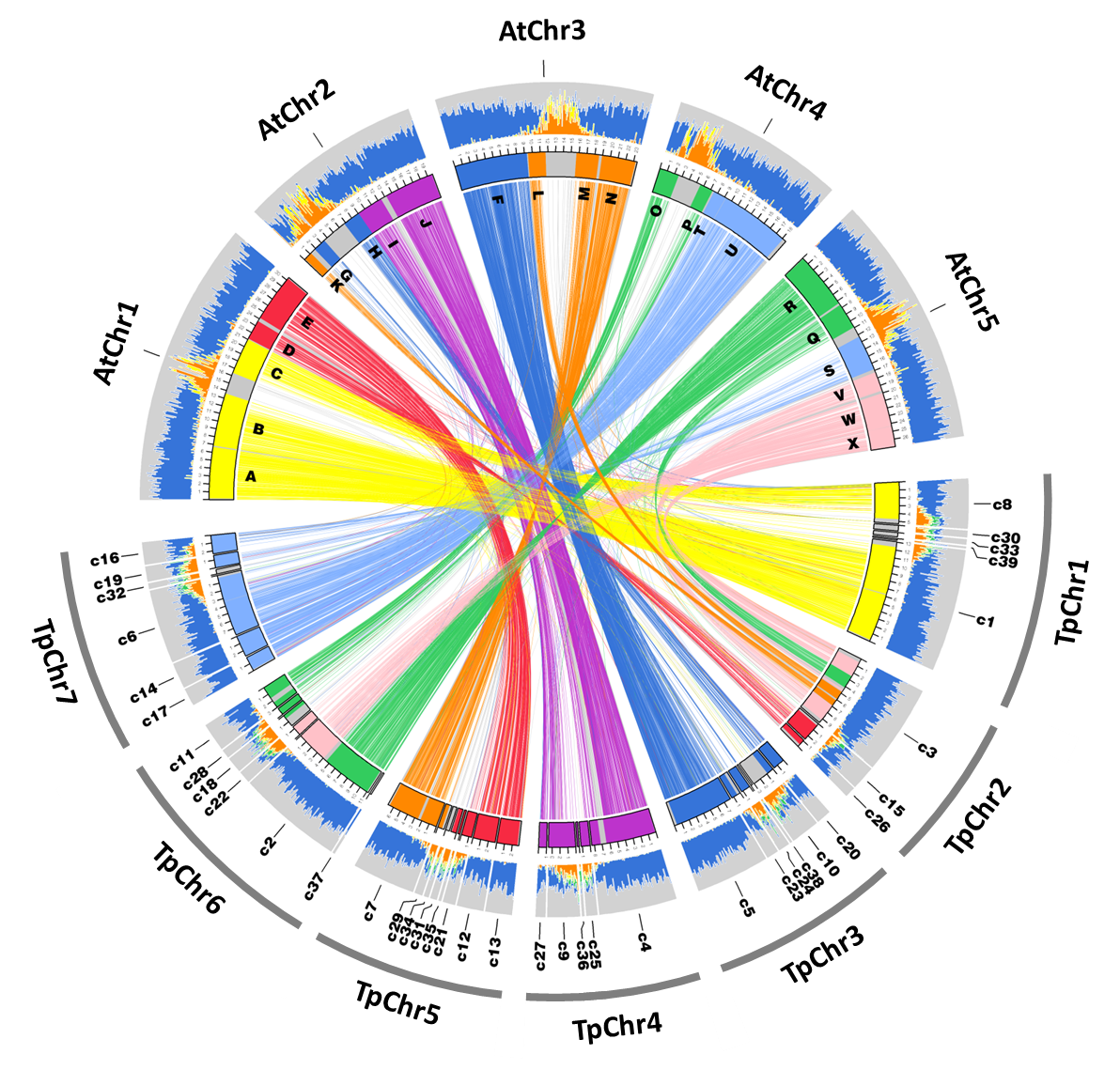
The first version of the S. parvula assembly has contigs assembled as seven scaffolds that represent seven chromosomes based on 454 and Illumina sequencing generated from a series of mate-pair and paired-end libraries as described in (Dassanayake et al., 2011). The assembly Version1, accompanying annotations that include coding and non-coding gene models and repeats, and the cleaned sequenced reads are available via NCBI BIOPROJECT PRJNA63667 and http://thellungiella.org/data/. The following update specifically refers to Version2.2 where the initial assembly was further curated by removing contigs that had significant redundancy and the annotation was refined to include only protein coding gene models.
Assembly and Annotation Statistics
-
Assembly-Annotation version: 2.2
-
Total contig length / count: 120,016,289 bp / 928
-
Contig L50 / N50: 6,763,654 bp / 7
-
Contig L90 / N90: 1,046,468 bp/ 27
-
Total number of scaffolds: 7 (each represents a total of 7 chromosomes)
-
Total number of protein coding transcripts/genes: 26,847
-
Number of contigs assigned to a chromosome: 37
-
Number of genes assigned to a chromosome: 25,093
-
Number of contigs without chromosome assignments: 891
-
Number of genes assigned to contigs not included in chromosomes: 1,754
-
Median gene length: 1,792 bp
-
Average number of exons per gene: 5.43
-
Median exon length: 140 bp
-
Median intron length: 105 bp
-
Number of genes with an Arabidopsis ortholog: 19,110 (based on 1-to-1 reciprocal best homologs)
-
Number of genes with a functional annotation: 22,711 (GO annotation)
-
Number of lineage specific genes: 2,912 (without an A. thaliana homolog); 547 (without a homolog in both A. thaliana and Eutrema salsugineum)
-
% of BUSCO units: 97.1% Complete (95.1% Single-copy and 2.0% Duplicated), 1.2% Fragmented, and 1.7% missing out of a total of 1,375 BUSCOs defined in embryophyte ortholog database v10
Sequencing, assembly, gene prediction, and annotation
The genotype used for the reference genome was derived from a single plant propagated from single seeds over eight successive generations. The original accession was collected from the shores of the hypersaline lake, Tuz in central Turkey at an elevation of 905 m above sea level. The use of a diploid genotype was confirmed via flow cytometry and karyotype analysis prior to sequencing (Oh et al., 2010).
Library construction and sequencing were performed in the W.M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign. Mate-pair, paired-end, and single-end random shotgun genomic libraries that included 3, 8, and 20 kb insert sizes were constructed for 454-GS FLX Titanium and Illumina GA2 platforms. A combined total of 7.8 Gb sequences with an average read size of 355 bp (454) and 80 bp (Illumina) were generated for the assembly. Approximately 85% of all sequences were derived from 454 sequencing. We followed a custom pipeline for a hybrid assembly starting from primary contigs generated by Newbler (454-Roche) and ABySS (Simpson et al., 2009) used with Nucmer (Kurtz et al., 2004), minimus2 (Sommer et al., 2007), and Vmatch tools (Kurtz, 2017) as described in the genome release (Dassanayake et al., 2011). Briefly, the raw reads were iteratively assembled to get the best possible contigs. Contigs that contained more than four genes unambiguously co-linear to their A. thaliana homologs in an Ancestral Crucifer Karyotype block (Mandáková & Lysak, 2008) were scaffolded to pseudomolecules representing chromosomes. The current assembly Version V2 has been created by further removing redundant contigs. This version has been used as the reference S. parvula genome in multiple comparative genomic studies since 2013 (Haudry et al., 2013; Oh et al., 2014; Oh & Dassanayake, 2019).
The assembly Version 1.0 was first masked for repeats using RepeatMasker (Jurka et al., 2005) for the prediction of gene models. Gene predictions were primarily generated using FGENESH++ (SoftBerry) and GENESCAN (Burge & Karlin, 1997). When the predictions from the two programs deviated for the same genomic region, the ORF closest in length to another known homologous cDNA was taken as the preferred gene model. The corresponding protein-coding gene model annotation for Version 2.0 was created with transcript model predictions made by Stringtie (Pertea et al., 2015) using additional RNA-seq data generated subsequently. High-confidence transcripts were selected to improve pre-existing protein-coding gene models in Version 1.0; fix chimeric and truncated gene models that did not agree with RNA-seq data; as well as to add UTRs when possible to create the Version 2.2 protein-coding gene models. Therefore, the Version 2.2 annotation includes protein coding gene models predicted in Version 1.0 further refined using RNA-seq data. Currently, only the primary protein-coding transcript models are included representing one transcript per locus. All predicted ORFs were searched against NCBI nucleotide and protein databases and TAIR9 cDNA database using BLASTn and BLASTx searches followed by the assignment of GO annotations to provide the most informative set of annotations possible.
Contributors:
Maheshi Dassanayake (LSU), Dong-Ha Oh (LSU), Hans J Bohnert (Univ. of Illinois), Jeffrey S Haas (Univ. of Illinois), Alvaro Hernandez (Univ. of Illinois), Hyewon Hong (Gyeongsang National Univ., Korea), Shahjahan Ali (KAUST), Dae-Jin Yun (Konkuk Univ., Korea), John M Cheeseman (Univ. of Illinois), Guannan Wang (LSU), Pramod Pantha (LSU), Kieu-Nga Tran (LSU), and Chathura Wijesinghege (LSU).
Funding for the genome project:
The S. parvula genome project was initially funded by the World Class University Program (R32–10148) at Gyeongsang National University, Republic of Korea; the Next-generation BioGreen21 Program (SSAC, PJ008025), Rural Development Administration, Republic of Korea; and a collaborative grant from the King Abdullah University of Science and Technology, Thuwal, Saudi Arabia, awarded to Hans J. Bohnert (University of Illinois at Urbana-Champaign, Urbana, IL). Additional support was provided by Dae-Jin Yun, Ray A Bressan, and Jian-Kang Zhu (Purdue University, West Lafayette, IN). Recent and ongoing updates to the genome have been supported by the National Science Foundation awards MCB 1616827 and EDGE 1923589 to Maheshi Dassanayake (Louisiana State University, Baton Rouge, LA).
Acknowledgments:
Keck Center for Comparative and Functional Genomics, University of Illinois at Urbana-Champaign and High Performance Computing at Louisiana State University (HPC@LSU)
References if citing the genome:
-
Dassanayake M, Oh D-H, Haas JS, Hernandez A, Hong H, Ali S, Yun D-J, Bressan RA, Zhu J-K, Bohnert HJ, et al. 2011. The genome of the extremophile crucifer Thellungiella parvula. Nature Genetics 43: 913–918.
-
Oh D-H, Hong H, Lee SY, Yun D-J, Bohnert HJ, Dassanayake M. 2014b. Genome structures and transcriptomes signify niche adaptation for the multi-ion tolerant extremophyte Schrenkiella parvula. Plant Physiol 164: 2123–2138.
Contacts
-
Maheshi Dassanayake (Louisiana State University) (email: maheshid@lsu.edu)
-
Dong-Ha Oh (Louisiana State University) (email: ohdongha@lsu.edu)
References used in this overview
-
Burge C, Karlin S. 1997. Prediction of complete gene structures in human genomic DNA11Edited by F. E. Cohen. Journal of Molecular Biology 268: 78–94.
-
Dassanayake M, Oh D-H, Haas JS, Hernandez A, Hong H, Ali S, Yun D-J, Bressan RA, Zhu J-K, Bohnert HJ, et al. 2011. The genome of the extremophile crucifer Thellungiella parvula. Nature Genetics 43: 913–918.
-
Dittami SM, Tonon T. 2012. Genomes of extremophile crucifers: new platforms for comparative genomics and beyond. Genome Biol 13: 166.
-
Hajiboland R, Bahrami-Rad S, Akhani H, Poschenrieder C. 2018. Salt tolerance mechanisms in three Irano-Turanian Brassicaceae halophytes relatives of Arabidopsis thaliana. Journal of Plant Research.
-
Haudry A, Platts AAE, Vello E, Hoen DRD, Leclercq M, Williamson RJ, Forczek E, Joly-Lopez Z, Steffen JG, Hazzouri KM, et al. 2013. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat Genet 45: 891–898.
-
Jurka J, Kapitonov V V, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research 110: 462–467.
-
Kazachkova Y, Eshel G, Pantha P, Cheeseman JM, Dassanayake M, Barak S. 2018. Halophytism: What Have We Learnt From <em>Arabidopsis thaliana</em> Relative Model Systems? Plant Physiology 178: 972 LP – 988.
-
Kiefer M, Schmickl R, German D a., Mandáková T, Lysak MA, Al-Shehbaz IA, Franzke A, Mummenhoff K, Stamatakis A, Koch MA. 2014. BrassiBase: Introduction to a novel knowledge database on brassicaceae evolution. Plant Cell Physiol 55: 1–9.
-
Koenig D, Weigel D. 2015. Beyond the thale: comparative genomics and genetics of Arabidopsis relatives. Nature reviews. Genetics 16: 285–298.
-
Kurtz S. 2017. The Vmatch large scale sequence analysis software.
-
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biology 5: R12.
-
Mandáková T, Lysak MA. 2008. Chromosomal phylogeny and karyotype evolution in x=7 crucifer species (Brassicaceae). Plant Cell 20: 2559–2570.
-
Oh D-H, Dassanayake M. 2019. Landscape of gene transposition–duplication within the Brassicaceae family. DNA Research.
-
Oh D, Dassanayake M, Bohnert HJ, Cheeseman JM. 2012. Life at the extreme: lessons from the genome. Genome Biology 13: 241.
-
Oh D-H, Dassanayake M, Haas JS, Kropornika A, Wright C, D’Urzo MP, Hong H, Ali S, Hernandez A, Lambert GM, et al. 2010. Genome structures and halophyte-specific gene expression of the extremophile Thellungiella parvula in comparison with Thellungiella salsuginea (Thellungiella halophila) and Arabidopsis. Plant Physiol 154: 1040–1052.
-
Oh D-H, Dassanayake M, Hong H, George S, Paeng SK, Kropornika A, Bressan RA, Lee S., Yun D-J, Bohnert HJ. 2014. Genomics of plant abiotic stress tolerance.
-
Orsini F, D’Urzo MP, Inan G, Serra S, Oh D-H, Mickelbart M V, Consiglio F, Li X, Jeong JC, Yun D-J, et al. 2010. A comparative study of salt tolerance parameters in 11 wild relatives of Arabidopsis thaliana. J Exp Bot 61: 3787–3798.
-
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotech 33.
-
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome research 19: 1117—1123.
-
Sommer DD, Delcher AL, Salzberg SL, Pop M. 2007. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8: 64.
-
The Plant List. 2013. http://www.theplantlist.org/
-
Uzilday B, Ozgur R, Sekmen AH, Yildiztugay E, Turkan I. 2015. Changes in the alternative electron sinks and antioxidant defence in chloroplasts of the extreme halophyte Eutrema parvulum (Thellungiella parvula) under salinity. Annals of Botany 115: 449–463.
-
Wang G, Pantha P, Tran K-N, Oh D-H, Dassanayake M. 2019. Plant Growth and Agrobacterium-mediated Floral-dip Transformation of the Extremophyte Schrenkiella parvula. JoVE (Journal of Visualized Experiments): e58544.
-
Whited J. 2015. The Next Top Models. Cell 163: 18–20.